Electronic Structure
Diabatic States for Electron and Energy Transfer
While electronic structure has proved remarkable successful in recent years at constructing adiabatic electronic eigenstates for ever larger molecules, these techniques always assume frozen nuclei. When nuclear-electronic coupling is non-negligible, however, these adiabatic electronic states are not necessarily stable. A classic example would be Fe+2/Fe+3 complexes, where the adiabatic states predict complete charge delocalization over the two metal centers. It is known, of course, that charge does localize and, as such, the adiabatic eigenstates are not stable or meta-stable in solution. We have recently developed tools that calculate diabatic states by "localizing" a set of adiabatic eigenstates, much in the same spirit as quantum chemistry has localized orbitals for the past 50 years (since Ruedenberg's famous 1963 paper.) Using these localized states as guesses for the initial and final states of electron and energy transfer processes, we have already modeled some nonequilibirum processes very accurately within the context of Marcus theory. Work is ongoing for how to find the optimal diabatization method and how to best integrate these methods with electronic structure theory. We have shown that these methods produce diabatic states with very small derivative couplings.
12. Subotnik, Yeganeh, Cave and Ratner. Constructing Diabatic States from Adiabatic States: Extending Generalized Mulliken Hush to Multiple Charge Centers with Boys Localization J. Chem. Phys. 129, 244101 (2008) [PDF] | TYPO's-[pdf]
16. Subotnik, Cave, Steele and Shenvi. The Initial and Final States of Electron and Energy Transfer Processes: Diabatic States as Motivated by System-Solvent Interactions J. Chem. Phys. 130, 234102 (2009) [PDF] | TYPO's-[pdf]
33. Fatehi, Alguire, and Subotnik. Derivative couplings and analytic gradients for diabatic states, with an implementation for Boys-localized configuration-interaction singles. J. Chem. Phys. 139, 124112 (2013) [PDF] | TYPO's-[pdf]
Accurate Excited State Energies
Calculating ground state energies is standard practice in the world of quantum chemistry, but calculating excited state energies is far more difficult. In particular, one requires a reasonably high degree of accuracy if one wants to accumulate the correct ordering of excited states, and also obtain the correct geometries for excited state crossings. Our efforts at generating accurate excited state energies have been based upon the idea of adding "post CIS" corrections for electron-electron correlation. In particular, we are guided by the principle of adding doubles corrections on top of CIS wavefunctions. Thus far, we have had some successes, but challenges do remain in balancing the energies of charge transfer states, locally excited states, Rydberg states, and the ground state.
32. Liu, Ou, Alguire and Subotnik. Communication: An inexpensive, variational, almost black-box, almost size- consistent correction to configuration interaction singles for valence excited states. J. Chem. Phys, 138, 221105 (2013) [PDF]
38. Liu and Subotnik. The Variationally Orbital-Adapted Configuration Interaction Singles (VOA-CIS) Approach to Electronically Excited States. [PDF]
Derivative Couplings and TD-DFT
From the perspective of standard, adiabatic quantum chemistry, all non-Born Oppenheimer effects are incorporated in the so-called derivative couplings. One of our group's research interests is the construction of derivative couplings between excited states, where we have found several interesting themes (and many more still exist).
26. Fatehi, Alguire, Shao and Subotnik. Analytical Derivative Couplings Between Configuration Interaction Singles States with Built-in Translation Factors for Translational Invariance
J. Chem. Phys. 135, 234105 (2011)
[PDF] Link
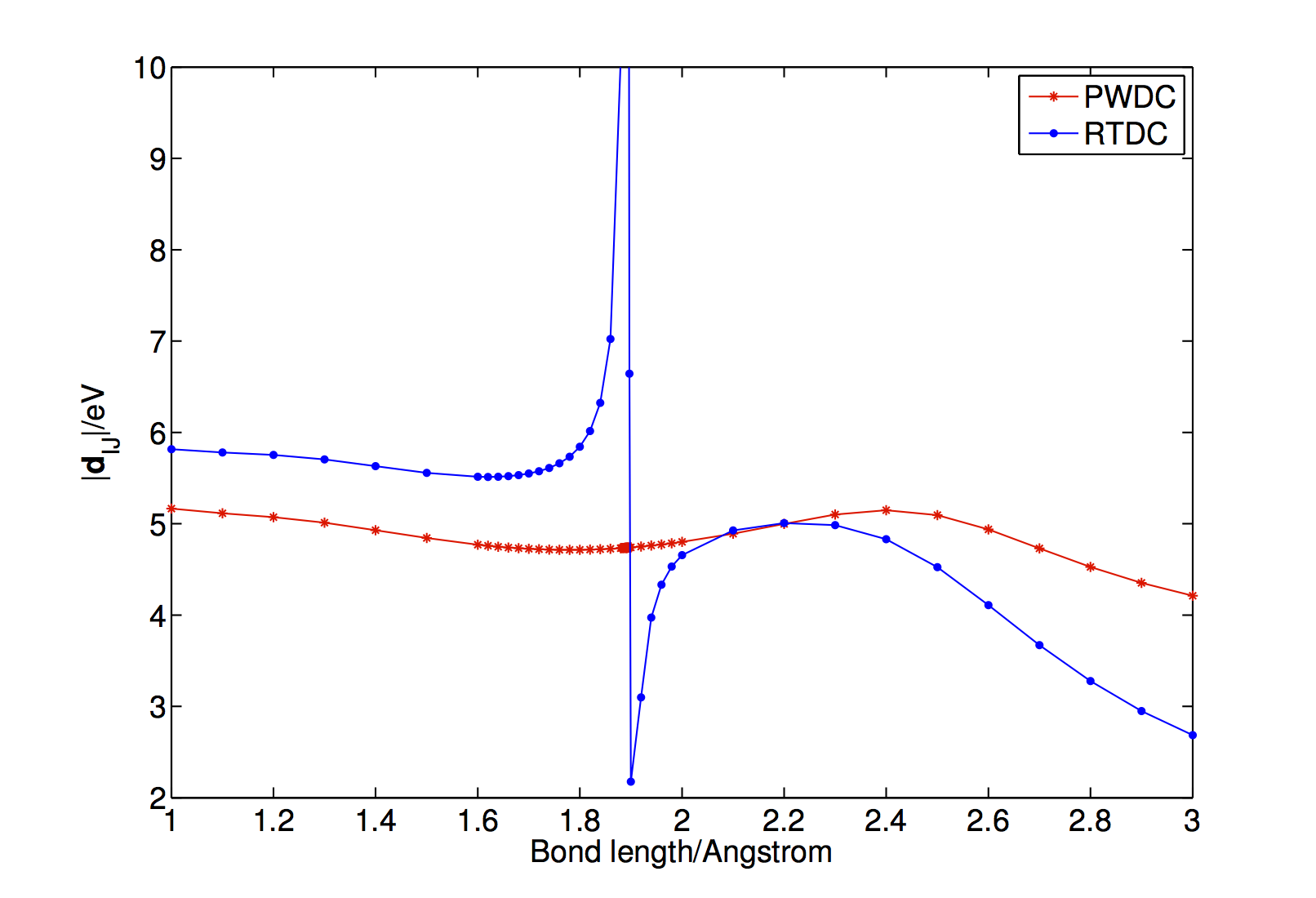
J. Phys. Chem. B 119, 7140-7149 (2015) [PDF] link
J. Phys. Chem. B 119, 7150-7161 (2015) [PDF] link
J. Chem. Phys. 142 064114 (2015) [PDF] Link